Papain-like cysteine peptidases (PLPs) are found in all domains of life. They degrade proteins by hydrolysis of peptide bonds. In animals, they are called cysteine cathepsins and are located primarily in lysosomes. Active cysteine cathepsins are monomeric, single-domain proteins. The active site is located at the top of the molecule and contains a Cys-His catalytic diad.
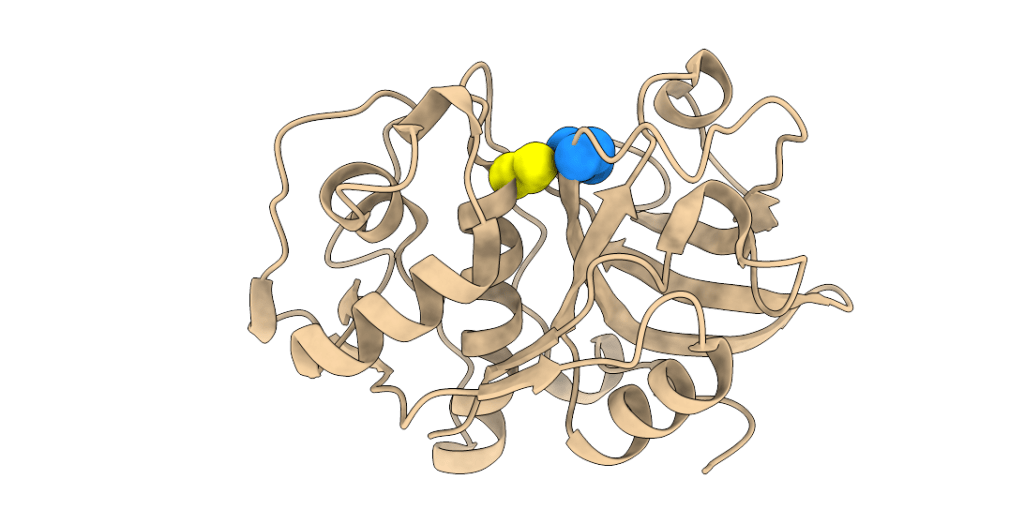
Three-dimensional structure of a papain-like peptidase. Cys and His residues of the catalytic diad are shown as yellow and blue spheres, respectively.
The principal and best-known endogenous regulators of cysteine cathepsins are proteinaceous inhibitors that bind into the active site. However, allosteric regulation has emerged in recent years as an important mode of regulation in vivo and targeting sites outside of the active site is a promising strategy in drug development. The most studied cysteine cathepsin in this respect is cathepsin K, an important peptidase in bone resorption by osteoclasts. Based on current knowledge we believe that proteinaceous inhibitors are emergency inhibitors that prevent unwanted proteolysis in the wrong place, whereas allosteric mechanisms regulate the proteolysis of specific protein substrates.
Our work
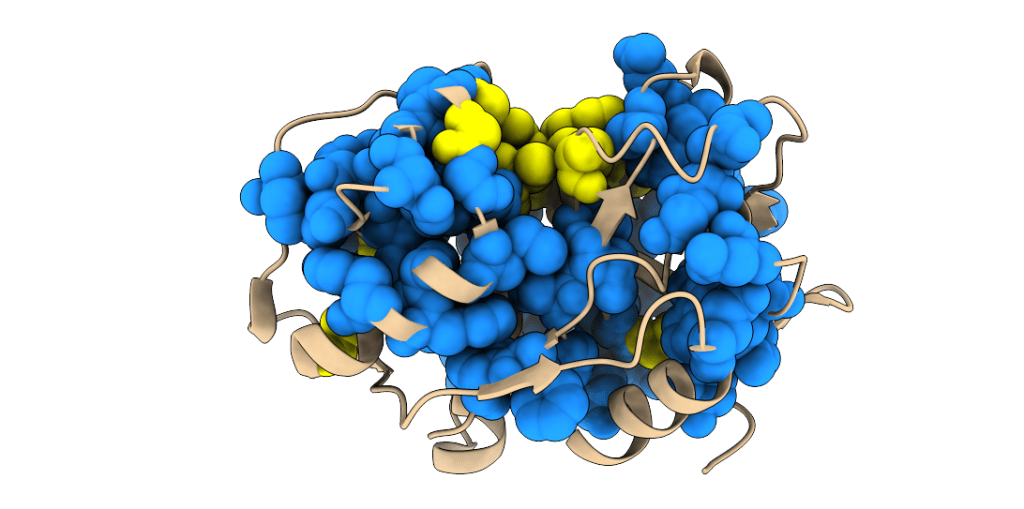
Our research is aimed at identifying and characterising molecular mechanisms of allosteric regulation in PLPs and is based on a combination of experimental and computational methods. As a starting point we investigated evolutionarily conserved mechanisms of allosteric regulation in PLPs using the statistical coupling analysis (Lockless & Ranganathan. Science. 1999. 286:295–299). We identified the presence of one protein sector in these enzymes (shown as blue spheres in the image below). The protein sector, as defined by Halabi et al. & Ranganathan (Cell. 2009. 138(4):774-86), is a network of residues that transmits allosteric communication between the active site and distant regulatory (allosteric) sites. Initial work was done on human cathepsin K as the model enzyme, but we have since expanded our work to other PLPs.
The protein sector (blue) and highly conserved residues (active site, yellow) of papain-like peptidases, illustrated on the structure of human cathepsin K.
Multiple allosteric sites are present on the surface of PLPs. We characterized a prototype allosteric binding site for small molecule effectors on cathepsin K that is located on the bottom-right side of the molecule.
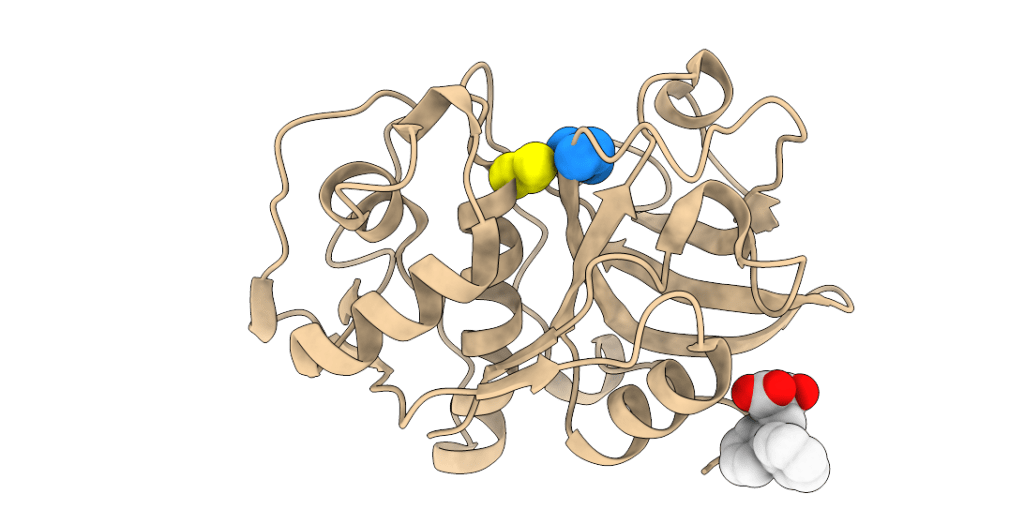
Crystal structure of effector NSC94914 (white spheres) bound to cathepsin K.
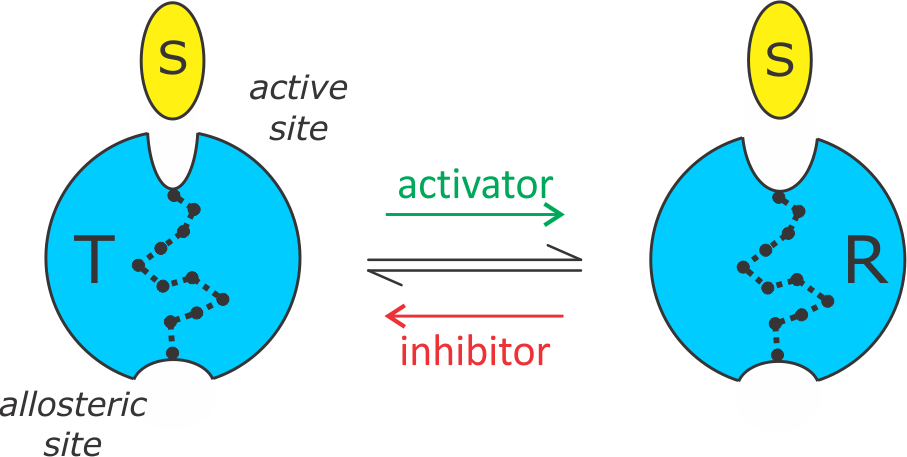
By our current interpretation, allosteric regulation in PLPs can be adequately described by a simple two-state model. The enzyme exists in equilibrium between a low-affinity T state and high-affinity R state. Allosteric inhibitors shift the equilibrium towards the T state and activators shift it towards the R state.
Allosteric two-state model.
Structurally, the two states differ in the width of the active site cleft in its narrowest region (S1 and S2 subsites) that binds the P1 and P2 residues of the substrate. Allosteric inhibitors stabilize a narrow conformation of the S1-S2 cleft (T state), whereas allosteric activators stabilize a wide conformation of the cleft (R state). We proposed this model initially for cathepsin K, where multiple allosteric sites are known. MD simulations indicate that the same mechanism applies for all allosteric sites. Evidence suggests that other PLPs operate by the same mechanism.
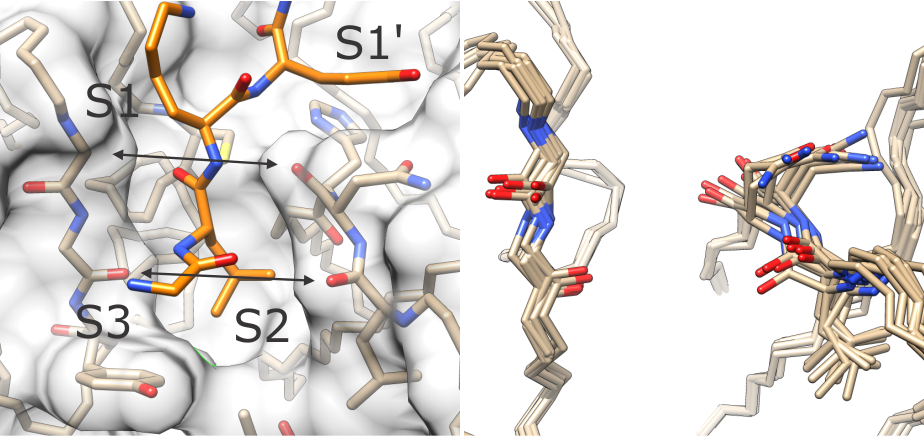
(left) The T and R states differ in the width of the active site cleft around subsites S1 and S2. (right) Ensemble of conformations of loops lining the S1-S2 cleft in human cathepsin K.
Future challenges
– identify communication pathways that transmit allosteric communication,
– discover the evolutionary diversity and conservation of allosteric mechanisms in PLPs,
– develop optimized allosteric effectors for use in vitro and in vivo.


