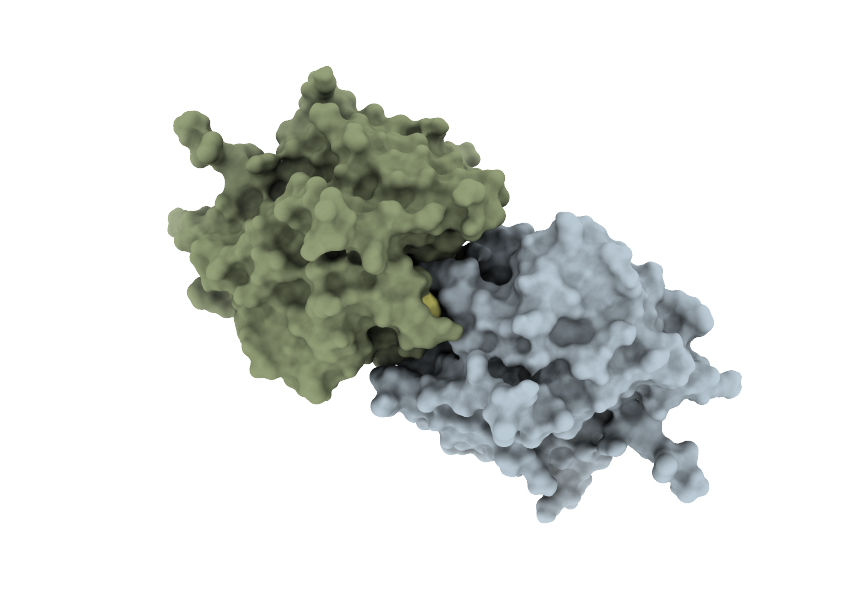
COOPERATE – Engineering of cooperative peptidases
In this project we aim to engineer oligomeric cooperative variants of papain-like cysteine proteases using a combination of rational computer-aided design and experimental screening of mutant protein libraries. The project is sponsored by ARIS (grant no. N1-0211).
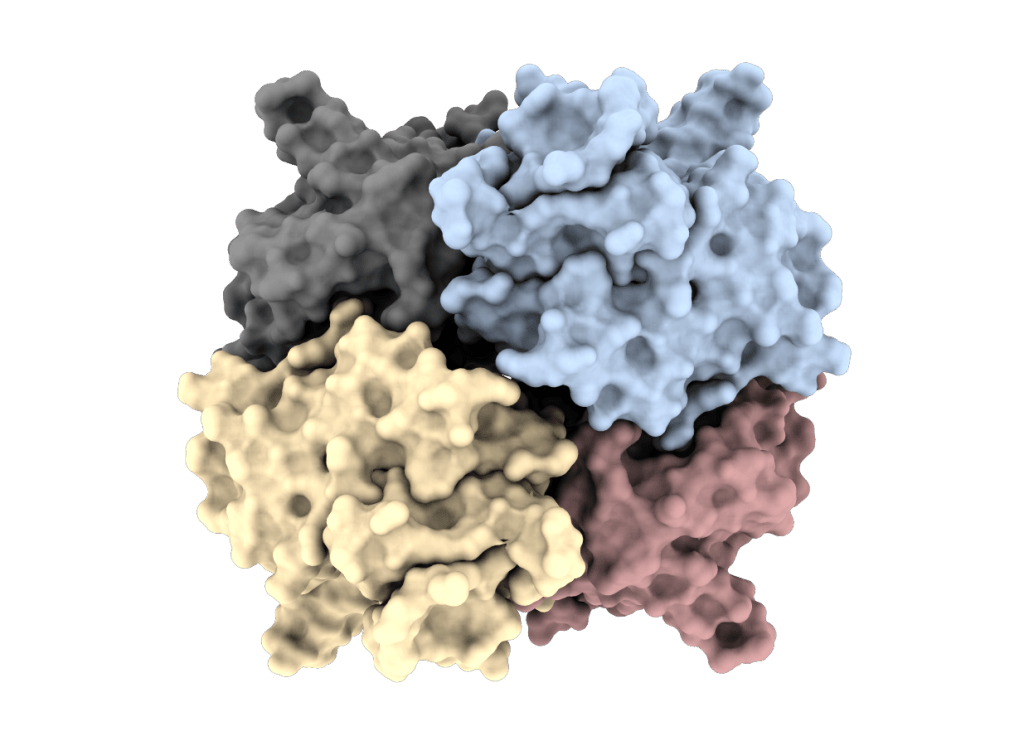
Oligomeric states and evolution of DPPI
In close connection with project COOPERATE, we aim to determine the diversity of oligomeric states and dynamics of oligomeric transitions incathepsins C from humans and other eukaryotes.
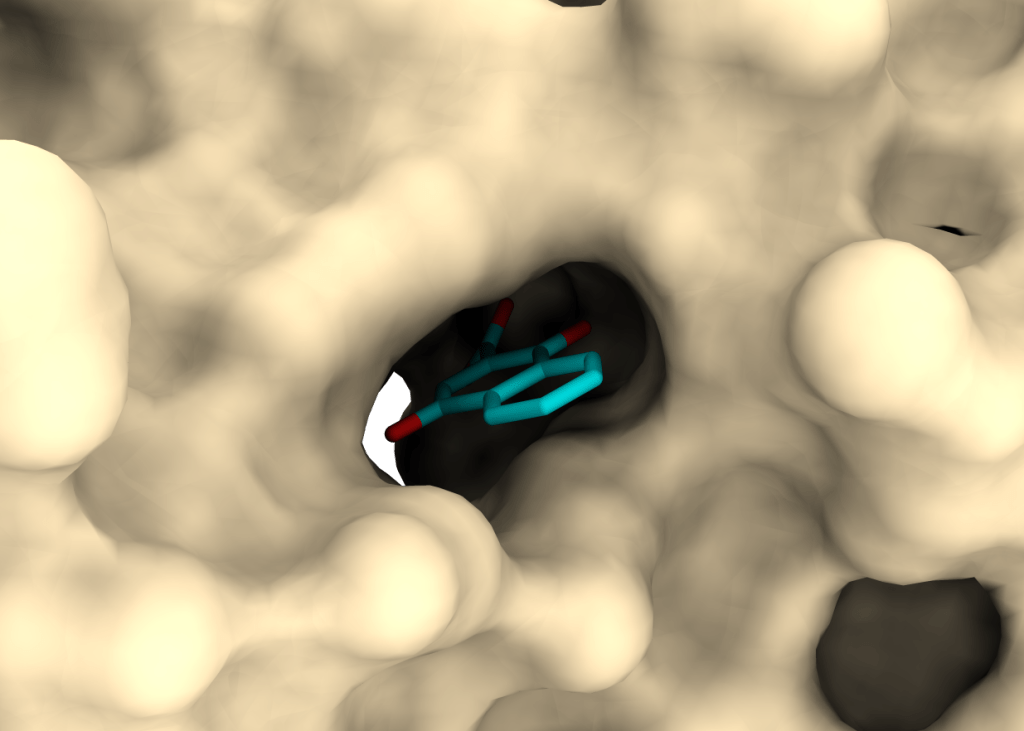
PRISM – PRotein Interactions with Small Molecules
The acronym PRISM stands for a long-term goal of our group to identify novel bioactive compounds synthesized at UL FKKT. The major emphasis is on identifying compounds with antimicrobial activity and their molecular (protein) targets.
Allosteric regulation of papain-like peptidases
Our major research topic for over a decade that originates from my postdoctoral stay in the lab of prof. Antonio Baici at Uni Zurich. We characterized glycosaminoglycans as the first known allosteric effectors of these peptidases and identified first small molecule allosteric effectors. We also investigated the structural basis for transmission of allosteric communication.



